Introduction
A major goal of drug discovery is to identify bioactive molecules that modulate molecular processes.
Rational drug design uses SAR analysis to link chemical structure with biological activity, while QSAR builds predictive models to estimate potency and effects.
These models guide compound design and optimization, improving efficacy, minimizing side effects, and ensuring new drugs offer more benefits than risks before approval.
Analyzing these patterns is challenging due to the large volume of data and features involved.
Selecting features for a QSAR model is complex and resource-intensive, so artificial intelligence is crucial for handling large datasets.
A robust AI-based QSAR model also requires high-quality data with variables that influence drug design.
- Structure variables: Shear rate, particle diameters…
- Particle features variables: Adhesive strength, resistance…
- ADME properties: absorption, distribution, metabolism, and excretion. In some cases, other properties like toxicity can be used.
After building the data set, you would need to analyze that data and build the model to design or optimize rational drugs.
Benefits
Below, you can see the benefits that these machine learning techniques can provide in drug designing processes:
![]()
![]()
Discover structure patterns
Study the surface properties, molecular volumes, or molecular interactions.
![]()
![]()
Identify behaviour influences
Relate the orientation of the molecule to its characteristics.
![]()
![]()
Anticipate the charasteristics
Develop a predictive model capable of predicting the behavior of a molecule in accordance with its design.
![]()
![]()
Improve drug design
Get better results and design better medicines while reducing costs.
Artificial intelligence is crucial in drug discovery, with neural networks outperforming traditional methods in SAR/QSAR modeling.
They predict biological activity, manage complex relationships, reduce costs, and identify key molecular descriptors.
Although expertise is usually needed, user-friendly tools like Neural Designer make AI methods accessible, allowing data analysis and model validation without programming.
It can be applied to drug design, healthcare, engineering, banking, and retail.
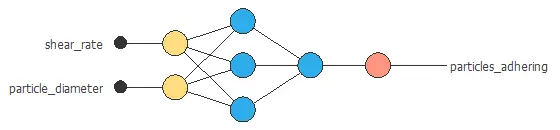
This article concludes that by using machine learning, you will be able to produce better results in less time while also being more cost-efficient.



